This is a demo store. No orders will be fulfilled.
 首页
首页 400-620-6333
400-620-6333
PCR、MIQE指南
背景:
目前,对于如何最好地执行和解释定量实时PCR(qPCR)实验缺乏共识。许多出版物中缺乏足够的实验细节,这阻碍了读者批判性评估所呈现结果的质量或重复实验的能力,从而说明了这个问题。
内容:
定量实时PCR实验(MIQE)指南发布的最低信息旨在确保结果的可靠性,以帮助确保科学文献的完整性,促进实验室之间的一致性,并提高实验透明度。MIQE是一组指南,描述了评估qPCR实验所需的最小信息。其中包括一份清单,随手稿首次提交给出版商。通过提供所有相关的实验条件和分析特征,评审员可以评估所用方案的有效性。为了使其他研究人员能够重现结果,有必要全面披露所有试剂、序列和分析方法。MIQE详细信息应以缩写形式或在线补充形式发布。
总结:
遵循这些指南将鼓励更好的实验实践,允许对qPCR结果进行更可靠和明确的解释。
基于荧光的定量实时PCR(qPCR)15(1-3)能够检测和测量来自多种来源的大量样本中的微量核酸,是分子诊断、生命科学、农业和医学的卓越技术(4,5)。其概念和实用的简单性,以及在均匀分析中的速度、灵敏度和特异性的结合,使其成为核酸定量的试金石。除了用作研究工具外,还开发了许多诊断应用,包括微生物定量、基因剂量测定、转基因食品中转基因的鉴定、癌症复发的风险评估和法医应用(6-11)。
这种受欢迎程度反映在大量报告qPCR数据的出版物中,这些出版物总是使用不同的试剂、协议、分析方法和报告格式。在如何最好地执行qPCR实验方面,这种明显的缺乏共识的情况产生了一系列严重缺陷的不利后果,这些缺陷阻碍了其作为独立标准的地位(12)。影响分析性能的技术缺陷包括:(a)样品储存、制备和核酸质量不足,产生高度可变的结果;(b) 逆转录引物和PCR引物和探针的选择不当,导致效率低下和不稳定的检测性能;(c)不适当的数据和统计分析,产生的结果可能具有高度误导性。因此,存在着科学文献被大量报告不充分和相互冲突结果的出版物所破坏的真正危险(13)。科学“2005年突破”报告的发表(14)和撤回(15)提供了一个令人不安的警告。由于缺乏使用该技术的大多数研究报告的特征信息,许多出版物没有提供足够的实验细节,使读者无法批判性地评估所提供结果的质量或重复实验,这一问题更加严重。具体来说,有关样本采集和处理、RNA质量和完整性、逆转录细节、PCR效率和分析参数的信息经常被忽略,而样本归一化通常是针对单个参考基因进行的,没有充分的理由。
本文件旨在为作者、审核人和编辑提供表1中规定的最低信息规范,qPCR实验必须报告这些信息,以确保其相关性、准确性、正确解释和可重复性。MIQE(定量实时PCR实验出版的最低信息,发音为mykee)是基于为DNA微阵列分析(16)、蛋白质组学实验(17)、基因组序列规范(18)以及正在讨论的RNA干扰工作(19,20)和代谢组学(21)制定的类似指南建模的,所有这些都是在MIBBI(生物和生物医学研究的最低信息,http://www.mibbi.org) (22 ). 虽然预计这些准则的未来更新可能会包括这样一项建议,但未提议强制纳入允许数据共享的通用报告语言。相反,这些指南的目标是结果的可靠性,以帮助确保科学文献的完整性,促进实验室之间的一致性,并增加实验透明度。阅读时应结合最近的出版物,深入探讨qPCR标准化问题(23-26)。
1.术语
一些术语需要标准化以确保澄清:
1.1我们建议缩写qPCR用于定量实时PCR,RT-qPCR用于逆转录-qPCR。将缩写RT-PCR应用于qPCR会引起混淆,并且与传统(传统)逆转录-PCR的使用不一致。
1.2用于标准化的基因应称为参考基因,而不是看家基因。
1.3TaqMan探针应称为水解探针。
1.4术语FRET探针(荧光共振能量转移探针)是指一种通用机制,其中发射/猝灭依赖于2个荧光染料分子的电子激发态之间的相互作用。LightCycler型探针应称为双杂交探针。
1.5《牛津英语词典》只列出了量化,而没有列出量化;因此,前者是恰当的词。
1.6描述用于定量的分数PCR循环的术语与文献中当前使用的阈值循环(Ct)、交叉点(Cp)和起飞点(TOP)不一致。这些术语都是指实时仪器的相同价值,是由实时仪器的竞争制造商出于产品差异而创造的,而不是科学准确性或清晰度。根据RDML(实时PCR数据标记语言)数据标准(http://www.RDML.org)(27),我们建议使用量化周期(Cq)。
2.概念考虑
为了解释和证明指南,我们发现回顾围绕qPCR实验的一些关键问题很有用:
2.1分析灵敏度是指样本中可通过分析准确测量的最小拷贝数,而临床灵敏度是指具有给定缺陷且分析确定为该条件阳性的个体百分比。通常,灵敏度表示为检测限(LOD),即可以通过给定的分析程序合理确定(通常使用95%的概率)检测到的浓度。假设泊松分布,理论上可能的最敏感LOD为每个PCR(28)3个拷贝,在PCR中包含至少1个拷贝的95%几率,以及单拷贝检测。实验程序通常包括样品处理步骤(即提取)和必要时的反转录。如果考虑了体积变化和这些步骤的效率,理论上可能最敏感的LOD可以用与实验相关的单位表示,例如每纳克组织的拷贝数。不得报告低于理论可能LOD的实验结果。也就是说,“0”的结果是没有意义和误导性的。qPCR分析中的LOD估计因Cq的对数性质而复杂,因为当模板浓度为零时,Cq是未定义的。qPCR中LOD的适当确定和建模是继续研究的重点(26)。
2.2分析特异性是指qPCR分析检测适当的靶序列,而不是样本中也存在的其他非特异性靶。诊断特异性是指没有特定条件的个体的百分比,其检测结果为该条件的阴性。
2.3准确度是指实验测量浓度和实际浓度之间的差异,以倍数变化或拷贝数估计值表示。
2.4重复性(短期精密度或批内方差)是指在同一分析中重复分析相同样本的分析的精密度和稳健性。它可以表示为Cq方差的SD。或者,可以使用拷贝数或浓度方差的SD或CV。然而,CV不应与Cqs一起使用(29)。
2.5再现性(长期精度或内部方差)是指运行之间或不同实验室之间结果的变化,通常表示为拷贝数或浓度的SD或CV。不同运行产生的Cq值受固有运行间变化的影响(30);因此,报告运行间Cq变化是不合适的。
描述靶基因mRNA浓度的出版物应明确目标是什么。大多数人类基因和其他多细胞生物中的许多基因的转录本是可变剪接的(31,32),这些剪接变体指定了替代蛋白质亚型,剪接模式在不同组织或不同发育阶段发生变化。因此,基于单个外显子的RT-qPCR分析可能检测到许多剪接变体,而跨内含子的引物可能更具选择性,但可能会遗漏一些剪接变体。最近,已经描述了在表达中显示等位基因不平衡的常染色体非印迹基因(33)。综上所述,这些发现意味着仅针对mRNA的一个或最多2个外显子的RT-qPCR分析不再足以描述特定基因的表达水平。因此,必须提供引物的序列信息,并评估其对转录本和单核苷酸多态性数据库中记录的已知剪接变体和单核苷酸多态性位置的特异性。对于从RTprimerDB数据库(34,35)中选择的引物集,可以通过查阅RTprimerDB网站轻松完成(http://www.rtprimerdb。org),其中包含所有相关信息。对于商业分析,需要批次信息和供应商的实验验证标准。强烈反对报告未经验证的商业分析和仅在电子版中验证的分析的结果。
必须记住,检测mRNA的存在并不能提供该mRNA是否会被翻译成蛋白质或功能性蛋白质是否被翻译的信息。
免疫组织化学、蛋白质印迹或其他蛋白质定量方法并不总是能够证实定量细胞mRNA数据。现已证实,mRNA和蛋白质浓度数据之间经常缺乏一致性(36),这对于指定作为多功能蛋白质复合物一部分的蛋白质的mRNA尤其如此(37)。最后,已经明确的是,了解特定微小RNA的存在和功能对于理解基因表达和量化mRNA物种同样重要(38)。
还需要注意的是,大多数定量RNA数据不是绝对的,而是相对的。因此,用于标准化的参考基因或材料至关重要,对RT-qPCR实验有效性的任何评估也必须考虑相对定量参考的适当性。因此,开发通用参考DNA和RNA校准材料虽然非常有用(39,40),但并不是万能的灵丹妙药(41,42)。
RT-qPCR实验中报告的表达值的大部分差异不仅仅是由于实验协议的变化,而是由各种数据处理算法应用的校正引起的,每个算法都对数据做出了自己的假设。因此,尽管qPCR经常被宣布为试金石或金标准,但在实践中,该“标准”是可变的,结果报告需要相当复杂的分析和解释(43)。
3.研究与诊断应用
qPCR技术的应用可大致分为研究应用和诊断应用。研究应用程序通常以相当低的吞吐量和许多不同的样本类型分析广泛的目标。需要解决的主要参数与分析灵敏度和特异性有关,在这种情况下,这分别是指分析可以检测多少靶拷贝以及无模板对照(NTC)是否可靠阴性。
相反,诊断应用程序通常分析有限数量的目标,但需要仅针对少数样本类型的高通量协议。虽然所有适用于研究应用的考虑因素也适用于诊断分析,但临床诊断分析有许多额外的要求需要考虑。这些要求包括有关分析灵敏度和特异性的信息,在这种情况下,这些信息是指当存在靶标时,分析结果呈阳性的频率,以及在没有靶标的情况下呈阴性的频率。此外,实验室内部和实验室之间的准确度和精密度通常由外部QC计划进行监测。其他临床实验室要求包括生成可报告结果的标准、是否对样本进行重复测量、关于假阳性/假阴性数据分辨率的数据以及使用相同和不同技术的多个实验室结果的相似性。到目前为止,只进行了几次实验室间比较,这两项研究都强调了定量PCR诊断分析标准化的必要性(44,45)。另一项实验室间工作计划在欧盟-罗佩恩框架7项目中进行:SPIDIA(体外诊断通用预分析工具和程序的标准化和改进;http://www.spidia.eu)。
4.样品采集、处理和制备
样本采集是实验可变性的第一个潜在来源,尤其是针对RNA的实验,因为mRNA图谱很容易受到样本采集和处理方法的干扰。有人建议,新鲜组织可以在冰上保存,而不会对RNA质量和浓度产生重大影响(46),但尽管这一假设可能适用于某些mRNA和组织,但可能并不普遍适用。因此,最好保持谨慎。因此,重要的是详细报告组织样本的获取位置以及是否立即处理。如果样品未立即处理,则有必要报告样品的预处理方式、储存时间和条件。
对样品的简要描述也很重要。例如,对肿瘤活组织检查的显微镜检查将揭示活组织检查中由肿瘤细胞组成的百分比,并且应报告该信息。
核酸提取是第二个关键步骤。提取效率取决于充分的同质化、样品类型(例如,原位组织与对数期培养细胞)、目标密度、生理状态(例如,健康、癌变或坏死)、遗传复杂性和处理的生物量。因此,有必要提供核酸提取方法的细节,并描述用于测量核酸浓度及其质量的方法。这些细节对于从新鲜冷冻激光显微切割活检样本中提取的RNA特别重要,因为组织制备程序的变化对RNA产量和质量都有重大影响(47)。
5.QC核酸
5.1.RNA样本
提取样本中RNA的定量非常重要,因为在比较不同样本时,建议使用大致相同数量的RNA。然而,有几种常用的定量过程,包括分光光度法(纳米滴;Thermo-Scientific)、微流控分析(安捷伦科技公司的生物分析仪、Bio-Rad实验室的Experion)、毛细管凝胶电泳(Qiagen的QIAxcel)或荧光染料检测(Am-bion/Applied Biosystems的核糖绿)。这些方法产生不同的结果,因此尝试比较用不同方法获得的数据是不明智的(48)。定量RNA的首选方法使用荧光RNA结合染料(如核糖绿),最适合检测低靶浓度。在任何情况下,建议仅用一种方法测量所有样品,并报告此信息。
检测和报告基因组DNA污染的程度,并记录可耐受的此类污染量的阈值截止标准,也很重要。必须报告RNA样品是否用DNase处理(包括使用的DNase类型和反应条件),并报告比较结果通过每个核酸靶点的阳性和非反向转录对照获得Cqs的子。
记录RNA模板的质量评估也很重要。该要求不适用的唯一情况是提取的总RNA数量太低,无法进行质量评估。当从单个细胞、血浆、其他无细胞体液、一些激光捕获的样本或澄清的组织培养基中提取RNA时,就会出现这种情况。它也适用于提取和RT-qPCR步骤作为连续单管实验执行的情况。报告的关键信息包括RNA数量、完整性以及是否存在逆转录或PCR抑制剂。值得一提的是,RNA在体内显著降解,这取决于对环境刺激反应的mRNA的自然调节(49)。这种RNA降解的来源超出了研究人员的控制范围;其表现之一是,即使是高质量的RNA样本也可以显示单个mRNA的差异降解。
A260/A280比率必须在中性pH下的缓冲液中测量,但如果要将核酸用于定量分析,尤其是当目的是测量细胞mRNA浓度的微小差异(<10倍)时,这种测量是不够的。吸光度比确实提供了RNA纯度的指示,因为DNA或残留苯酚的存在改变了该比率。相反,至少应该提供凝胶电泳证据,或者更好的是,提供基于微流体的rRNA分析(50)或参考基因/靶基因3:5'完整性分析(51)的结果。使用生物分析仪/Experion系统计算RNA完整性数或RNA质量指标数的优势在于,这些测量提供了有关RNA样本总体状态的定量信息。然而,重要的是要记住,这些数字与rRNA质量有关,不能期望成为质量的绝对衡量标准。使用3:5'分析要求两种分析的PCR效率几乎相同(51),并且不受差异抑制。该分析还需要建立阈值标准,以描述足以产生可靠结果的RNA质量。理想情况下,该分析应针对一组可能没有内含子的“完整性参考基因”,其3:5’阈值比约为0.2–5。显然,需要进一步的工作来生成一种普遍适用、经济高效且简单的用于评估RNA完整性的前组分。
应通过稀释样品(最好)或使用SPUD(52、53)等通用抑制试验来检查逆转录活性或PCR的抑制。如果RNA样本显示部分降解,则必须报告该信息,因为检测低水平转录物的分析灵敏度可能会降低,转录物降解的相对差异可能会产生错误的靶比。
5.2.DNA样本
一般来说,DNA的降解问题要小得多;然而,重要的是能够评估DNA降解的程度,以用于法医应用,即在犯罪现场或大规模灾难现场或涉及失踪人员案件的现场的恶劣环境条件可能会降解DNA的化学结构的情况下。qPCR分析的小扩增子大小有助于最大限度地减少与分析相关的问题,但已开发出可提供DNA质量定量测量的方法(54),应考虑用于此类专门目的。
抑制的可能性是一个更普遍适用的变量,必须在公共场合解决,重要的是确保与DNA共纯化的抑制剂不会扭曲结果,例如病原体检测及其量化(55)。Al-虽然可以使用这种方法,例如用阳性对照物(52)加标样本来检测抑制,但不同的PCR反应可能不同样容易受到核酸提取物中共价物质的抑制(56,57)。因此,最好常规使用核酸稀释液来证明Cqs或拷贝数的减少与预期结果一致,并报告这些数据。
6.反转录
逆转录步骤将大量变异引入RT-qPCR分析(58,59)。因此,有必要详细描述用于将RNA转化为cDNA的方案和试剂。该文件必须包括RNA反转录的数量、启动策略、酶类型、体积、温度和反转录步骤的持续时间。建议重复或三次执行反转录步骤,并且每个样本中的总RNA浓度相同(58)。
7.qPCR
qPCR分析必须提供以下信息:每个靶基因和参考基因的数据库登录号,每个引物和任何探针的外显子位置,每个寡核苷酸的序列和浓度,包括任何染料和/或修饰碱基的识别、位置和链接。还需要聚合酶的浓度和特性、每个反应中模板(DNA或cDNA)的数量、Mg2+浓度、缓冲液的确切化学成分(盐、pH值、添加剂)和反应体积。研究人员还必须确定他们使用的仪器,并记录所有PCR循环条件。由于使用的耗材会影响热循环,因此有必要确定单管、带材或板材的使用及其制造商。所用塑料制品的透明度(例如白色或透明)也很重要,因为不同的塑料在荧光反射和灵敏度方面表现出显著差异(60)。使用板材时,密封方法(热粘接与粘合剂)可能会影响板材周围样品的蒸发,因此应记录在案。
由于PCR效率高度依赖于所使用的引物,因此必须公布其序列。即使使用商用引物,这一要求也是完全可行的,因为公司有先例可以提供其引物和探针序列(http://www.primerdesign.co.uk/使用.asp)进行研究。
此外,强烈鼓励提交到公共数据库,如RTprimerDB;随着时间的推移,这些数据库可能成为全球信息交换所。
7.1.二级结构
核酸靶标的结构(例如,茎环二级RNA结构)对逆转录和PCR的效率有很大影响。因此,引物、探针和PCR扩增子的位置必须考虑RNA模板的折叠。应使用核酸折叠软件检查序列,例如DNA的mfold(http://mfold.bioinfo.rpi.edu/cgi-bin/dna-form1.cgi)或RNA(http://frontend.bioinfo.rpi.edu/applications/mfold/cgi-bin/rna-form1-2.3.cgi)。理想情况下,折叠结构应提供给审查人员。
7.2.特异性
在电子工具中,如BLAST或等效特异性搜索对于分析设计很有用。应记录与假基因或其他意外靶点的任何明显同源性,并作为对齐序列提供以供审查;然而,特异性必须通过直接实验证据(电泳凝胶、熔融曲线、DNA测序、扩增子大小和/或限制性内切酶消化)进行验证。
预测寡核苷酸熔化温度(Tm)的算法对于初始设计很有用,但退火的实际最佳温度必须通过实验确定。虽然引物优化已不流行,但很明显,退火优化不佳对分析质量有很大影响(51)。引物二聚体的显著存在会降低基于探针的分析中的PCR效率,并可能在基于SYBR Green I的分析中产生假阳性。应向审查人员提供一些引物优化的证据,最好以退火温度或Mg2+梯度的形式提供,并以Cq值、荧光与周期数的曲线图和/或熔化曲线表示(61)。
7.3.控制和量化校准器
除了上述RT-qPCR分析中的no-反转录控制外,所有qPCR反应都需要额外的控制和/或定量校准品。当使用探针时,NTC检测PCR污染,还可以将未扩增的扩增产物(例如引物二聚体)与SYBR绿色反应中的预期PCR产物区分开来。NTC应包含在每个板或每批样品上,并且拒绝数据的条件应为
已建立。例如,如果未知最低浓度的Cq为35,则Cqs为2:40的NTC可以忽略。
从实验样品中提取的核酸形式的阳性对照有助于监测随时间变化的分析,并且在每次运行中没有执行校准曲线时是必不可少的。
定量校准品可以是纯化的靶分子,例如跨越完整PCR扩增子的合成RNA或DNA寡核苷酸、质粒DNA构建物、克隆到质粒的cDNA、体外转录的RNA、参考RNA池、特定生物样品的RNA或DNA,或国际公认的生物标准(如可用)。应将稀释液稀释至规定浓度的载体tRNA(酵母或大肠杆菌,浓度为10–100纳克/微升)。为了检测人类病原体,可以将校准品稀释到阴性对照样品RNA或DNA中,也可以将其稀释到健康人血浆中,然后可以在载体tRNA存在下进行裂解。特定模板的连续稀释液可作为储备溶液制备,以抵抗多次冻融循环。当检测到Cq偏移为0.5–1.0时,应准备新批次。或者,校准曲线的溶液可以在4°C下储存一周,然后丢弃。
对于诊断分析,定量PCR应包括一个独立验证的校准品(如果可用),该校准品位于分析的线性区间内。还建议使用阳性和阴性提取对照。
7.4.分析性能
必须确定以下检测性能特征:PCR效率、线性动态范围、LOD和精度。
7.4.1.PCR效率。稳健和精确的qPCR检测通常与高PCR效率相关。当报告靶基因相对于参考基因的mRNA浓度时,PCR效率尤其重要。LlLlCq方法是确定样本之间浓度差异的最常用方法之一,基于单个参考基因的正常化。计算目标基因和参考基因之间的Cq值差异(LlCq),并直接比较不同样本的LlCq。这两个基因必须以相当的效率进行扩增,才能准确地进行比较。然而,最流行的方法并不一定是最合适的,并且已经开发出了替代的、更通用的定量模型,以纠正扩增效率的差异(62),并允许使用多个参考基因(30)。
PCR扩增效率必须通过校准曲线来确定,因为这种校准提供了简单、快速和可重复的平均PCR效率、分析灵敏度和分析稳健性指标。放大效率应根据校准曲线对数线性部分的斜率确定。具体来说,当初始模板浓度(自变量)的对数绘制在x轴上,Cq(因变量)绘制在y轴上时,PCR效率=10-1/斜率-1。理论最大值为1.00(或100%),表明每次循环的产物量加倍。理想情况下,根据复制校准曲线报告估算PCR效率方法的CI或SE。
每个量化目标的校准曲线必须包含在提交的手稿中,以便审查人员可以获得这些信息;出版物中必须包括从这些校准曲线得出的斜率和y截距。PCR效率的差异将产生具有不同斜率的校准曲线。因此,由于模板量可变,目标物和参考物的Cq值之间的差异将不会保持恒定,相对浓度的计算将不准确,产生误导性结果。
Cq值>40是可疑的,因为这意味着效率低,通常不应报告;然而,使用这种任意Cq截止值并不理想,因为它们可能太低(消除有效结果)或太高(增加假阳性结果)(26)。
7.4.2.线性动态范围。必须描述反应线性的动态范围(通过校准曲线确定的最高到最低可量化拷贝数)。根据用于生成校准曲线的模板,动态范围应涵盖至少3个数量级,理想情况下应扩展到5或6 log10浓度。校准曲线的线性区间必须包括被量化的目标核酸的区间。由于定量下限通常定义不明确,因此应确定声称在线性区间内的最低浓度的变化。必须报告相关系数(r2值),理想情况下,应在整个线性动态范围内提供CI。
7.4.3.LOD。LOD定义为检测95%阳性样本的最低浓度。换句话说,在LOD浓度下含有目标的一组复制品中,失败反应不应超过5%。低拷贝PCR受随机限制,每个PCR的LOD不可能小于3拷贝。然而,如果形成多个反应,则可以通过数字PCR(29、63、64)获得较低浓度的准确定量。事实上,可以通过限制稀释来检查浓度校准器,以表明失败和成功反应的百分比遵循泊松分布。
7.4.4.精度。qPCR结果中的变化有很多解释,包括影响退火和/或去自然完成的温度差异、移液误差引起的浓度差异和随机变化。qPCR的精度通常随浓度而变化,随拷贝数而降低。理想情况下,分析内变异(重复性)应在图中显示为SD误差条或具有重复样本的校准曲线上的CI。CV不应与Cqs(29)一起使用,但可用于表示拷贝数或浓度的方差。这种技术变异应该与生物变异区分开来。生物复制可以直接解决各组或治疗之间qPCR结果差异的统计显著性。对于诊断分析,可能还需要报告站点和不同操作员之间的批间精密度(再现性)。
7.5.多路qPCR
多路复用的能力大大扩展了qPCR分析的能力(65,66),特别是当应用于同时检测点突变或多态射时(67)。多路复用要求提供证据,证明单管中多个目标的准确定量不会受到影响,即分析效率和LOD与以单管方式运行分析时相同。当丰度明显较低的靶点与丰度较高的靶点同时出现时,这一问题尤为重要。
8.数据分析
数据分析包括检查原始数据,评估其质量和可靠性,以及生成可报告的结果。描述了各种数据收集和处理策略,系统评估表明,qPCR数据分析方法在性能上有很大差异(68)。
关于数据分析和置信度估计方法的详细信息以及所用软件的规范是必要的。必须指定识别异常值的方法和此类数据的处理。记录分析精度需要识别用于评估方差(例如95%CI)的统计方法,并呈现相应的浓度或Cq值。此类信息应包括可重复性和可再生产性数据(如可用)。如上所述,为Cqs重新报告CV是不合适的(29),因为Cqs总是比为拷贝数计算的CV低(因此可能误码)。必须提供关于治疗准确性所用方法的信息,包括组间报告差异的统计显著性。
8.1.规范化
标准化是可靠qPCR分析的重要组成部分,因为该过程控制提取率、逆转录率和扩增效率的变化,从而能够比较不同样本中的mRNA浓度。使用参考基因作为内部对照是使细胞mRNA数据正常化的最常见方法;然而,尽管参考基因的使用通常被认为是最合适的归一化策略(69),但它们的效用必须在实验上对特定组织或细胞类型和特定实验设计有效。不幸的是,尽管人们越来越意识到系统验证的重要性,尽管使用不适当的参考基因进行归一化的潜在高度误导效应已广为人知,但这些考虑仍然被广泛忽视(70)。因此,许多分子分析仍然包含标准化较差的qPCR数据。
标准化涉及报告相关基因的mRNA浓度与参考基因的mRNA浓度的比率。参考基因mRNA应稳定表达,其丰度应与样本中存在的mRNA总量密切相关。
除非研究人员为评审人员提供明确的证据,确认其在所述实验条件下的恒定表达,否则不能接受针对单个参考基因的标准化。必须通过实验确定参考基因的最佳数量和选择,并报告该方法(71-73)。
8.2.可变性
生物系统的固有变异性可能与组间的实验差异相匹敌或超过。当使用许多生物复制来增加实验的统计显著性时,通常会观察到这种变化。虽然生物逻辑复制之间的差异可能很大,但足够的数字可能允许识别较小的实验差异。最近的一份出版物提供了一个处理此类数据的教科书示例,以及如何从受高度生物变异影响的分析中挽救具有生物意义的数据(74)。许多因素导致实验变异,并影响达到给定统计功率所需的生物重复数。因此,功率分析有助于确定有效结论所需的样本数量。
8.3.定性分析
使用PCR仅检测核酸模板的存在,而不是准确定量,被称为定性PCR,广泛用于病原体诊断。PCR方法的定性/定量分层问题在于,准确的是/否答案需要有关PCR检测低端灵敏度的信息。因此,即使是定性分析也应提供有关分析性能特征的信息,尤其是第7.4.2节和第7.4.3节中讨论的要点。
结论
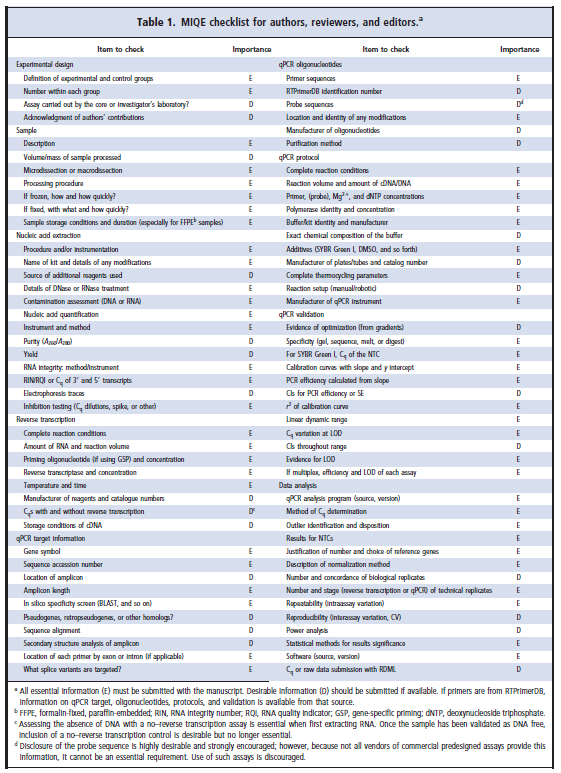
确保qPCR和RT-qPCR分析的质量保证措施的必要性已得到充分认可(25、44、75-86)。定量PCR和传统(传统)PCR检测之间的主要区别在于前者有可能准确定量靶核酸。必须清楚地识别这种差异,并且不能假设传统的PCR分析可以直接转换为qPCR格式。表1为准备qPCR研究报告的作者提供了检查表。被认为必要的项目(E)需要允许审查人员评估工作,并允许其他投资者复制工作。被认为可取的项目(D)也很重要,如有可能,应包括在内,但并非所有情况下都可用。当然,应用常识很重要:对于针对数百个目标的表达特征的初始筛选,没有必要遵守清单上的所有项目。然而,一旦确定了更有限的一组目标(少于20个),则应通过清单详细描述分析性能,该清单位于http://www.rdml.org/miqe/.
总之,这些指南的目的有三个方面:
1.使作者能够设计和报告具有更大内在价值的qPCR实验。
2.允许审稿人和编辑根据制定的标准衡量提交的手稿的技术质量。
3.为了更容易地复制遵循这些指南的已发表研究中描述的实验。
因此,使用这种广泛应用的技术的调查将产生更统一、更具可比性、最终更可靠的数据。
参考文献:
1.Higuchi R,Dollinger G,Walsh PS,Griffith R.Si-同时扩增和检测特定DNA序列。生物技术(纽约)1992;10:413–7.
2.Higuchi R,Fockler C,Dollinger G,Watson R.动力学PCR分析:实时监测DNA扩增反应。生物技术(纽约)1993年;11:1026 –30.
3.Wittwer CT,Herrmann MG,Moss AA,Rasmussen RP.快速循环DNA扩增的连续荧光监测。生物技术1997;22: 130–8.
4.Bustin SA。使用实时逆转录聚合酶链反应测定绝对定量mRNA。摩尔内分泌素2000;25:169 –93.
5.Kubista M,Andrade JM,Bengtsson M,Forootan A,Jonak J,Lind K,等。实时聚合酶链反应。Mol Aspects Med 2006;27:95– 125.
6.Bernard PS,Wittwer CT。癌症诊断的实时PCR技术。临床化学2002;48: 1178 – 85.
7.Mackay IM,Arden KE,Nitsche A.病毒学中的实时PCR。核酸研究2002;30:1292–305.
8.Mackay IM。微生物实验室中的实时PCR。临床微生物感染2004;10:190 – 212.
9.Bustin SA,Mueller R.实时反向转录PCR(qRT-PCR)及其在临床诊断中的潜在应用。临床Sci(Lond)2005;109: 365–79.
10.Bustin SA,Mueller R.实时反向转录PCR和结直肠癌隐匿性疾病的检测。Mol Aspects Med 2006;27: 192–223.
11.van den Berg RJ、Vaessen N、Endtz HP、Schulin T、van der Vorm ER、Kuijper EJ。在一项前瞻性多中心研究中评估实时PCR和常规诊断方法检测艰难梭菌相关性腹泻。医学微生物学杂志2007;56:36 – 42.
12.Bustin SA,Nolan T.定量实时逆转录聚合酶链反应的陷阱。J Biomol Tech 2004;15:155– 66.Garson JA,Huggett JF,Bustin SA,Pfaffl MW,Benes V,Vandesompele J,Shipley GL。多发性硬化症中人类内源性逆转录病毒-W(HERV-W)RNA表达和DNA拷贝数的不可靠实时PCR分析。艾滋病是逆转录病毒。即将于2009年推出。
14.Huang T,Bohlenius H,Eriksson S,Parcy F,Nilsson O.拟南芥基因FT的mRNA从叶片移动到茎尖并诱导开花。科学2005;309:1694 – 6.
15.Bohlenius H,Eriksson S,Parcy F,Nilsson O.撤回。科学2007;316:367.
16.Brazma A,Hingamp P,Quackenbush J,Sher-lock G,Spellman P,Stoeckert C等。关于微阵列实验(MIAME)的最小信息-朝向微阵列数据的标准。Nat Genet 2001;29:365–71.
17.Taylor CF,Paton NW,Lilley KS,Binz PA,Julian RK Jr,Jones AR,等。蛋白质组学实验的最低信息量(MIME)。Nat生物技术2007;25:887–93.
18.Field D,Garrity G,Gray T,Morrison N,Selengut J,Sterk P等。关于基因组序列(MIGS)规范的最小信息。Nat生物技术2008;26:541–7.
19.Echeverri CJ、Beachy PA、Baum B、Boutros M、Buchholz F、Chanda SK等。最大限度地降低大规模RNAi筛查中报告假阳性的风险。Nat方法2006;3:777–9.
20.Haney SA。提高RNAi筛选的稳健性和有效性。药物基因组学2007;8: 1037– 49.
21.Sansone SA、Fan T、Goodacre R、Griffin JL、Hardy NW、Kaddurah Daouk R等。代谢组学标准倡议。Nat Biotechnol 2007;25:846–8.
22.Taylor CF,Field D,Sansone SA,Aerts J,Apweiler R,Ashburner M等。促进生物和生物医学调查的一致最小报告指南:MIBBI项目。Nat Biotechnol 2008;26:889 –96.
23.Burns MJ,Valdivia H,Harris N.以转基因大豆定量为模型系统,分析和解释实时PCR跟踪检测方法的数据。Anal Bioanal Chem 2004;378: 1616 –23.
24.Burns MJ,Nixon GJ,Foy CA,Harris N.标准-实时定量PCR方法的数据处理-异常值评估和校准曲线比较。BMC Biotechnol 2005;5:31.
25.Ellison SL,English CA,Burns MJ,Keer JT。提高实时PCR低水平DNA分析可靠性的途径。BMC Biotechnol 2006;6:33.Burns MJ,Valdivia H.实时定量PCR检测极限建模。2008年《欧洲食品技术报告》;226:1513–24.
27.Lefever S,Hellemans J,Pattyn F,Przybylski DR,Taylor C,Geurts R等。RDML:实时定量PCR数据的结构化语言和报告指南。核酸研究[2009年2月17日出版前的Epub]。网址:http://nar.oxfordjournals.org/cgi/content/abstract/gkp056。
28.Wittwer CT,Kusakawa N.实时PCR。作者:Persing DH,Tenover FC,Versalovic J,Tang JW,Unger ER,Relman DA,White TJ,eds.分子微生物学:诊断原理与实践。华盛顿:ASM出版社;2004年,第71-84页。
29.Schmittgen TD,Livak KJ。通过比较CT方法分析实时PCR数据。Nat协议2008;3:1101– 8.
30.Hellemans J,Mortier G,De Paepe A,Speleman F,Vandesompele J.用于实时定量PCR数据管理和自动分析的qBase相对量化框架和软件。基因组生物学2007;8: R19。
31.de la Grange P,Dutertre M,Correa M,Auboeuf D.选择性剪接数据库的新进展:从目录到人类选择性剪接变体表达和功能调控的详细分析。BMC生物信息学2007;8:180.
32.Ben-Dov C,Hartmann B,Lundgren J,Valcarcel J.选择性前mRNA剪接的全基因组分析。生物化学杂志2008;283:1229 –33.
33.Bjornson HT,Albert TJ,Ladd Acosta CM,Green RD,Rongione MA,Middle CM,等。基于SNP特异性阵列的等位基因特异性表达分析。基因组研究2008;18:771–9.
34.Pattyn F,Robbrecht P,De Paepe A,Speleman F,Vandesompele J.RTPrimerDB:实时PCR引物和探针数据库,主要更新2006。核酸研究2006;34:D684-8。
35.Pattyn F,Speleman F,De Paepe A,Vandesompele J.RTPrimerDB:实时PCR引物和探针数据库。核酸研究2003;31: 122–3.Gygi SP,Rochon Y,Franza BR,Aebersold R.酵母中蛋白质和mRNA表达的相关性。摩尔细胞生物学1999;19:1720 –30.
37.Schmidt MW,Houseman A,Ivanov AR,Wolf DA。pombe裂殖酵母的比较蛋白质组学和转录组学分析。Mol Syst Biol 2007;3:79.
38.Landi D、Geminani F、Naccarati A、Pardini B、Vodicka P、Vodickova L等。微RNA结合位点内的多态性与spo-放射型结直肠癌的风险。致癌2008;29: 579 – 84.
39.Cronin M,Ghosh K,Sistare F,Quackenbush J,Vilker V,O'Connell C.基因表达的通用RNA参考材料。临床化学2004;50:1464 –71.Novoradovskaya N、Whitfield ML、Basehore LS、Novoradovsky A、Pesich R、Usary J等。作为微阵列实验标准的大学参考RNA。BMC基因组学2004;5:20.
41.Gingeras TR.基因表达研究的RNA参考材料。艰难的第一步。临床化学2004;50:1289 –90.
42.Joseph LJ。用于基因表达研究的RNA参考材料。RNA计量:预测需要部分清除。临床化学2004;50:1290 –2.
43.Bustin SA,Benes V,Nolan T,Pfaffl MW。定量实时RT-PCR-前景。摩尔内分泌杂志2005;34:597– 601.
44.Ramsden SC、Daly S、Geilenkeuser WJ、Duncan G、Hermitte F、Marubini E等。等量:实时PCR的国际外部质量评估方案。临床化学2006;52:1584 –91.
45.Damond F,Benard A,Ruelle J,Alabi A,Kupfer B,Gomes P等.人类免疫缺陷病毒2型(HIV-2)病毒载量定量分析的质量控制评估:2006年HIV-2感染国际合作的结果.临床微生物学杂志2008;46:2088 –91.Micke P,Ohshima M,Tahmasebpoor S,Ren ZP,Ostman A,Ponten F,Botling J.新鲜冷冻组织的生物银行:RNA在非固定手术标本中是稳定的。Lab Invest 2006;86:202–11.
47.Morrogh M、Olvera N、Bogomolniy F、Borgen PI、King TA。用于激光捕获显微切割和从新鲜冷冻乳腺组织中提取RNA的组织制备。2007年生物技术;43:41-2、4、6秒。
48.Bustin SA。实时荧光定量PCR:当前程序和偏好的快照。专家版Mol Diagn 2005;5: 493– 8.
49.Doma MK,Parker R.欧盟核生物中的RNA质量控制。Cell 2007;131:660 – 8.
50.Fleige S,Pfaffl MW。RNA完整性和对实时qRT-PCR性能的影响。Mol As-pects Med 2006;27:126 –39.
51.Nolan T,Hands RE,Bustin SA。使用实时RT-PCR定量mRNA。Nat协议2006;1:1559 – 82.
52.Nolan T,Hands RE,Ogunkolade BW,Bustin SA。SPUD:用于检测核酸制剂中抑制剂的qPCR分析。肛门生物化学2006;351:308 –10.蕨类植物RB,Garson JA。使用溴花叶病毒内部对照物开发和评估实时RT-PCR分析,以定量无细胞人类免疫缺陷病毒2型。J Virol Methods 2006;135:102– 8.
54.Swango KL,Hudlow WR,Timken MD,Buoncris-tiani先生。用于评估法医样本中核DNA数量和质量的多重qPCR分析的发育验证。法医科学国际2007;170:35– 45.
55.Garson JA,Grant PR,Ayliffe U,Ferns RB,Tedder RS.使用自动样本制备和小鼠巨细胞病毒内部对照对乙肝病毒DNA进行实时PCR定量。J Virol Methods 2005;126:207–13.
56.Huggett JF,Novak T,Garson JA,Green C,Morris-Jones SD,Miller RF,Zumla A.PCR反应对抑制剂的差异敏感性:一个重要且未被认识的现象。BMC Res Notes 2008;1:70.
57.Ståhlberg A,Aman P,Ridell B,Mostad P,Kubista M.通过比较K和入 免疫球蛋白轻链表达。临床化学2003;49:51–9.
58.Ståhlberg A,Hakanson J,Xian X,Semb H,Kubista M.mRNA定量中逆转录反应的性质。临床化学2004;50:509 –15.Ståhlberg A,Kubista M,Pfaffl M.基因表达分析中逆转录酶的比较。临床化学2004;50:1678 – 80.
60.Reiter M,Pfaffl M.板位置、板类型和密封系统对实时PCR结果的影响。生物技术2008;22: 824–8.
61.Ririe KM,Rasmussen RP,Wittwer CT。通过分析聚合酶链反应期间的DNA熔化曲线进行产品分化。Anal Bio-chem 1997;245:154 – 60.Pfaffl MW。实时RT-PCR中相对定量的新数学模型。核酸研究2001;29:E45。
63.Dube S,Qin J,Ramakrishnan R.在纳米流体装置上使用数字PCR对DNA样本中拷贝数变化进行数学分析。PLoS ONE 2008;3: e2876。
64.Vogelstein B,Kinzler KW。数字PCR。Proc Natl Acad Sci U S A 1999;96:9236 – 41.Elnifro EM、Ashshi AM、Cooper RJ、Klapper PE。多重PCR:优化及其在诊断病毒学中的应用。临床微生物学2000版;13: 559 –70.
66.Wittwer CT、Herrmann MG、Gundry CN、Elenitoba Johnson KS。实时多重PCR分析。方法2001;25:430 – 42.
67.Elenitoba-Johnson KS、Bohling SD、Wittwer CT、King TC。通过多色荧光法和荧光熔融曲线分析进行多重PCR。Nat Med 2001;7:249 –53.
68.Karlen Y,McNair A,Perseguers S,Mazza C,Mer-mod N.定量PCR的统计意义。BMC生物信息学2007;8:131.
69.Huggett J,Dheda K,Bustin S,Zumla A.实时RT-PCR标准化;策略和考虑。基因免疫2005;6:279 – 84.
70.Gutierrez L、Mauriat M、Guenin S、Pelloux J、Lefebvre JF、Louvet R等。缺乏参考基因的系统验证:植物中逆转录聚合酶链反应(RT-PCR)分析中的一个严重陷阱。植物生物技术杂志2008;6:609 –18.
71.Vandesompele J,De Preter K,Pattyn F,Poppe B,Van Roy N,De Paepe A,Speleman F.通过对多个内控基因进行几何平均,对实时定量RT-PCR数据进行准确归一化。基因组生物学2002;3: 研究0034。Pfaffl MW,Horgan GW,Dempfle L.相对表达软件工具(REST),用于实时PCR中相对表达结果的分组比较和统计分析。核酸研究2002;30:e36。
73.Andersen CL,Jensen JL,Orntoft TF。实时定量逆转录PCR数据的标准化:一种基于模型的方差估计方法,用于识别适合标准化的基因,应用于膀胱癌和结肠癌数据集。癌症研究2004;64:5245–50.
74.Willems E,Leyns L,Vandesompele J.独立生物复制的实时PCR基因表达数据的标准化。Anal Bio-chem 2008;379:127–9.
75.Apfalter P,Reischl U,Hammerschlag先生研究中的内部核酸扩增分析:在依赖结果之前需要多少质量控制?临床微生物学杂志2005;43:5835– 41.
76.Ciabatti I,Froiio A,Gatto F,Amaddeo D,Marchesi U.转基因检测实时PCR方法的内部验证和质量控制:一种实用方法。Dev Biol(巴塞尔)2006;126: 79 – 86; 讨论324-5。
77.de Cremoux P,Bieche I,Tran Perennou C,Vig-naud S,Boudou E,Asselain B,等。使用实时逆转录聚合酶链反应对人类乳腺肿瘤中激素依赖性基因表达进行实验室间质量控制。内分泌相关癌症2004;11:489 –95.
78.de Vries TJ,Fourkour A,Punt CJ,van de Locht LT,Wobbes T,van den Bosch S,等。黑色素瘤患者外周血中酪氨酸酶和MART-1转录物检测的再现性:使用实时定量RT-PCR的质量控制研究。英国癌症杂志1999;80:883–91.
79.Gabert J,Beillard E,van der Velden VH,Bi W,Grimwade D,Pallisgaard N,等。欧洲癌症防治项目“实时”定量逆转录酶-聚合酶链反应融合基因转录物检测白血病残余疾病的标准化和质量控制研究。白血病2003;17:2318 –57.
80.Lemmer K,Donoso Mantke O,Bae HG,Groen J,Drosten C,Niedrig M.登革热病毒感染PCR诊断中的外部质量控制评估。J Clin Virol 2004;30:291– 6.
81.Marubini E,Verderio P,Raggi CC,Pazzagli M,Orlando C.实时PCR外部质量控制产生的统计诊断。国际生物标记杂志2004;19:141– 6.
82.Raggi CC、Verderio P、Pazzagli M、Marubini E、Simi L、Pinzani P等。意大利基于Taq-Man探针实时PCR的定量分析外部质量控制计划。临床化学实验室医学2005;43:542– 8.
83.Sjoholm MI,Dillner J,Carlson J.评估新鲜和存档干血点DNA的质量和功能性,以及质量控制指南的建议。临床化学2007;53: 1401–7.
84.Wang X,Jia S,Meyer L,Xiang B,Chen LY,Jiang N,等。利用预杂交第三染色图像进行全面质量控制,可以通过cDNA微阵列进行准确的基因表达测量。BMC生物信息学2006;7:378.
85.Winters MA,Tan LB,Katzenstein DA,Merigan TC。逆转录酶-聚合酶链反应定量血浆人类免疫缺陷病毒1型RNA的生物变异和质量控制。临床微生物学杂志1993;31: 2960 – 6.
86.van der Velden VH、Panzer Grumayer ER、Cazza-niga G、Flohr T、Sutton R、Schrauder A等。在多中心环境下优化基于PCR的儿童急性淋巴细胞白血病最小残留疾病诊断。白血病2007;21:706 –13.
Categories: 实验方案(Protocols)